A New Hope: Preventing Spinal Muscular Atrophy Through Prenatal Intervention
In a groundbreaking medical achievement, a two-and-a-half-year-old girl became the first person to receive treatment for the rare and often devastating disorder known as spinal muscular atrophy (SMA) while still in the womb. Today, she is defying the odds, showing no signs of the disease.
Above: A fetal ultrasound, a non-invasive imaging procedure that uses high-frequency sound waves to create a real-time image of the fetus in the womb. Image courtesy of UT Southwestern Medical Center.
SMA is a genetic disorder that affects approximately one in every 6,000 babies, making it one of the most common genetic disorders in young children and a leading cause of infant mortality. The disease affects motor neurons in the spinal cord—nerve cells that control voluntary muscle movement.
The condition is usually caused by mutations in the survival motor neuron gene 1 (SMN1), which contains instructions for making the SMN protein, a key player in maintaining healthy motor neurons. In individuals without SMA, the SMN1 gene produces 85-90% of the body's SMN proteins, while the SMN2 gene, which closely resembles SMN1 and encodes various forms of SMN proteins, contributes 10-15%. When the SMN1 gene is mutated, the body cannot produce enough SMN protein, leading to loss of motor neurons in the spinal cord. As a result, muscles—especially in the thighs, back, shoulders, and hips—begin to weaken and shrink from disuse and lack of nerve stimulation.
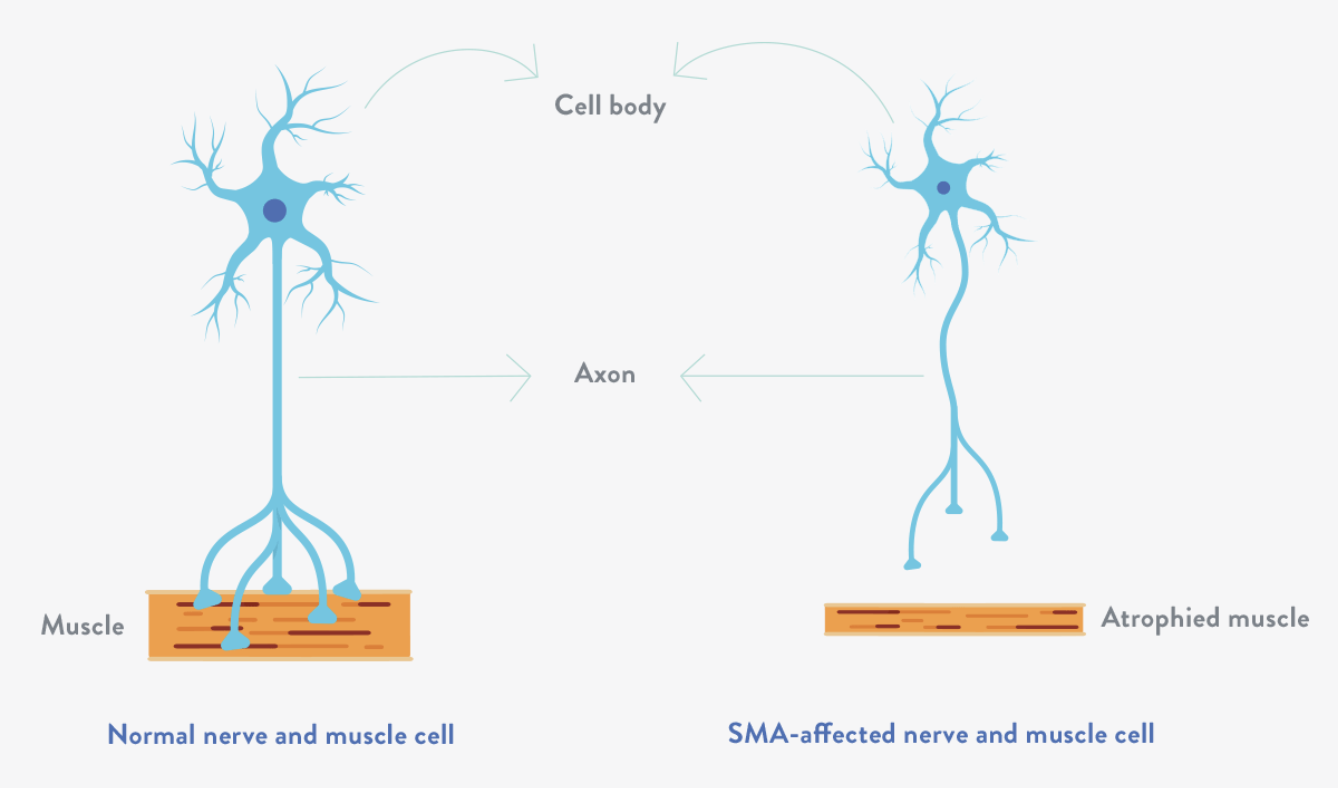
Above: Illustration showing the degeneration of motor neurons in SMA patients leading to the gradual decrease in the mass and strength of muscles (atrophy). Image courtesy of Together in SMA.
The severity of symptoms differs depending on the type of SMA a person has, with five types linked to SMN1 gene mutations. These types are classified by symptom severity and average age of onset. SMA type 1, which manifests within six months after birth, is the most common and severe form. Infants with SMA type 1 develop extreme weakness that causes difficulty breathing and swallowing, and they often die within the first few years of life without treatment or breathing support. A person can have multiple copies of the SMN2 gene, with more copies linked to the less severe forms. Extra copies of SMN2 can partially compensate for the SMN protein deficiency, though the amount of SMN protein produced by SMN2 is still significantly lower than what would be generated by a healthy SMN1 gene.
The idea of giving the drug in utero came from the parents who had already experienced the loss of one child with confirmed SMA type 1. When they conceived another child, genetic testing through amniocentesis revealed the fetus had no copies of the SMN1 gene (confirming an SMA diagnosis) and only two copies of the SMN2 gene, which is predictive of SMA type 1.
Risdiplam, one of three treatments approved by the U.S. Food and Drug Administration (FDA) for treating SMA in newborns, was selected for this groundbreaking study. Developed by a Swiss-based biotech company called Roche, risdiplam is an oral small-molecule drug that works by modulating the splicing of the SMN2 gene, effectively increasing SMN protein levels in SMA patients.
Above: Illustration of the chemical structure of risdiplam, the orally bioavailable mRNA splicing modifier used for the treatment of SMA. Image courtesy of PubChem.
While the use of risdiplam has shown promise, it has traditionally been administered only after birth due to limited knowledge of the drug’s in utero effects. In fact, risdiplam is generally not recommended in pregnant women due to animal data suggesting a risk of fetal harm. However, this timing presents a significant limitation, as research indicates that up to half of newborns with the most severe form of SMA already display symptoms at birth. Recognizing this critical treatment window, both the FDA and the local institutional review board approved this unprecedented single-patient investigational plan to administer treatment prenatally, potentially changing the course of the disease before irreversible damage could occur.
Researchers administered risdiplam orally to the mother at a dose of five milligrams daily from 32 weeks and five days post-conception until delivery. Weekly monitoring assessed the mother's obstetric health and potential drug-related side effects, while ultrasonography tracked fetal growth, activity, and anatomical development. Starting eight days after birth, the infant began receiving oral risdiplam, which has continued daily to the present (the child was 30 months old as of February 2025).
Testing of the amniotic fluid and both maternal and infant blood at birth revealed increased SMN levels and reduced neurofilament levels, indicating that the drug successfully modulated its target in utero. To date, the now two-and-a-half-year-old has displayed no features of SMA, such as hypotonia, weakness, areflexia, or fasciculation. Doctors have performed motor function, muscle ultrasonographic, and electrophysiological studies every six months, which have shown normal peripheral-nerve and muscle development for the infant’s age. While preliminary, the results of this case open the door for larger studies to see whether the findings can be replicated. Additionally, this breakthrough raises the possibility of treating other genetic conditions in utero when treatment after birth is insufficient.
