Authors: Emily Da Cruz*, Mohamad A. Mikati*+
*Division of Pediatric Neurology and Developmental Medicine, Department of Pediatrics, Duke University
+Department of Neurobiology, Duke University
Corresponding Author:
Mohamad A. Mikati, MD
Wilburt C. Davison Professor of Pediatrics
Professor of Neurobiology
Chief, Division of Pediatric Neurology and Developmental Medicine
Duke University
2301 Erwin Rd.
Durham, NC 27710
Phone: 919-668-4073
Fax: 919-681-8943
mohamad.mikati@dm.duke.edu
Highlights
- Review of iPSC differentiation methods for various neuronal subtypes
- Allows scientists to quickly reference various protocols that they can replicate in their own work
- Brief analysis comparing the efficiency of different methods, highlighting gaps in the current literature about iPSC differentiation
Abstract
Induced pluripotent stem cells (iPSCs) have been demonstrated to be a valuable tool in modeling diseases, investigating drug mechanisms, and understanding manifestations of disease in specific cell types. In the field of neurology, studies using iPSC are also investigating the use of iPSCs as a regenerative therapy, which could be extremely beneficial for understanding and treating neurodegenerative diseases. While iPSCs have been used in various studies, a lack of awareness of all available protocols for differentiation makes it difficult to identify best practices. In this article, methods of differentiating iPSCs into various neuron subtypes are described in order to provide a consolidated list of protocols that will aid the analysis of suitable and available methods for specific research protocols. By compiling and summarizing these various methods, future studies can determine which methods are best for their specific research goals.
Keywords: stem cells; iPSC; cell differentiation; neurons
I. Introduction
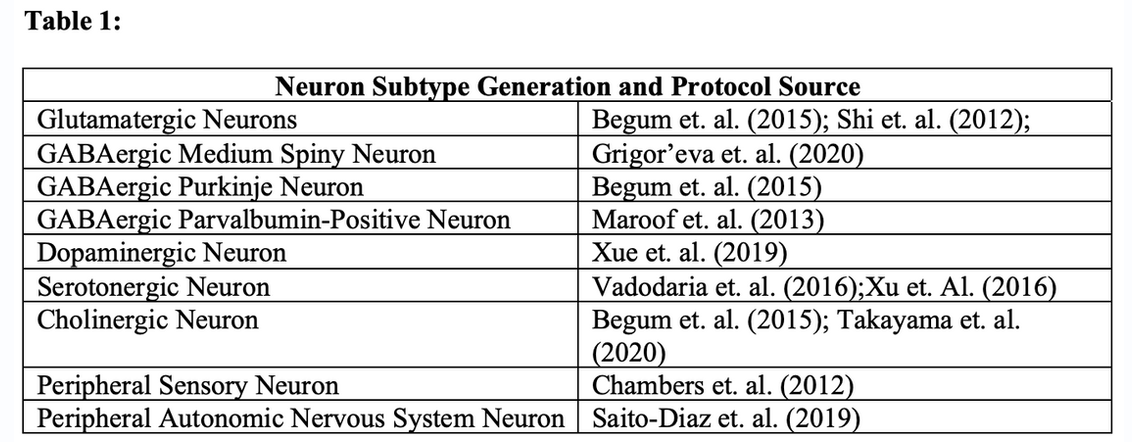
Cell manipulation has been an increasingly prominent technique used to better understand the mechanisms of diseases, pathways of drug actions, and effects of gene therapies (Ferrari et al., 2020, Freel et al., 2020). Induced pluripotent stem cells (iPSCs) are a uniquely useful tool for cell manipulation, as they are pluripotent stem cells that can be generated from somatic cells. iPSCs have been particularly vital to in-depth learning about various underlying mechanisms of disease through studying the cellular physiology in the cells with the patient’s genetic background. In the field of neurology, this is particularly useful since neuronal tissue from humans is not accessible for study as other tissues like blood are. Additionally, there is promise that iPSCs can be used as a regenerative therapy to modify local pathophysiology in the brain and to screen potential drugs for neurological disorders. While the use of iPSCs show promise for the future, a lack of a consolidated compilation of the protocols used to differentiate iPSCs into various neuronal types can lead to difficulties in planning for studies or in missing useful alternative methods. In this article, we review and present the differentiation methods used for generating various neuron subtypes, both in written and table form (see Table 1). We hope that by compiling and briefly presenting these various methods, future studies can determine which methods are best for their specific research goals
II. Historical Background
iPSCs were first generated in 2006 by Kazutoshi Takahashi and Shinya Yamanaka using four transcription factors, Oct3/4, Sox2, c-Myc, and Klf4 (now coined “Yamanaka factors”), in mouse embryonic or adult fibroblasts (Takahashi and Yamanaka, 2006). These iPSCs, with the ability to differentiate into an array of cell subtypes in vitro and in vivo, have expanded the control of pluripotency and changed the manner in which cell types can be analyzed and studied. With all four Yamanaka factors, iPSCs are both identical in pluripotency stem cells but are tumorigenic. The discovery by Nagawaka et. al. (2008) that the tumorigenicity could be reduced by removing the c-MYc transcription factor was instrumental in making iPSCs safer and practical for today’s clinical applications. Through differentiation of iPSCs into various cell types, studies regarding disease mechanisms, regenerative cell therapies, and drug mechanisms have been and continue to be completed (Aoi et. al., 2016). iPSC-based disease modeling has been particularly useful for genetic disease research, as patient-derived iPSCs can be used to study the impacts of genetic mutations on various cell types (Shi et. al., 2017). Furthermore, the potential use of iPSCs in regenerative therapies has been particularly important in the studies of Parkinson’s disease, spinal cord injuries, liver dysfunction, and more (Aoi et. al., 2016). Despite the immense progress that has been made in the biomedical field thus far due to iPSCs, there are still many barriers to be overcome before they can be applied to a clinical setting, as there has been a lack of consistent quality and streamlined differentiation protocols and materials that prevents the work done thus far from transferring from the lab to the clinic (Liu et. al., 2020). Thus, a review, like ours, that compiles and presents these methods would be helpful.
III. Neuron Subtype Generation from iPSCs
1) Glutamatergic
Begum’s methods of differentiation: Two different methods have been used in the past to differentiate iPSCs into glutamatergic neurons. The first method utilizes neurospheres (also known as progenitor cells) as an intermediate between iPSCs and glutamatergic neurons. Once iPSC colonies are formed, they are treated with collagenase IV to initiate neural differentiation. After being harvested and resuspended in a knockout serum replacement medium, the culture is plated on low adhesion suspension culture plate in a broth for approximately 3 days in order to generate the neurospheres, which are maintained in neuronal induction medium (NIM). The neurospheres are then collected and broken down into fragments on Matrigel-coated plates, leading to the formation of a neuroepithelial sheet known as a “neurosphederm.” This phase is followed by a 3–5-day incubation period which allows neural progenitors to form, thus preparing the culture for sub-type and region specific differentiation. Neuronal Maintenance Medium (NMM) was used in the plates until glutamatergic neurons were generated (roughly one week after neuronal induction). Begum et. al. states that this method has proved to be more efficient than pre-existing ones, specifically the neuroectoderm method and studies utilizing this method showed a doubling in expression of neuroprogenitor genes (Begum et. al. 2015).
Shi’s method of differentiation: Alternatively, Shi et. al. (2012) provides an extensive protocol to generate glutamatergic neurons from iPSCs. To start, iPSCs are cultured by plating mouse embryonic fibroblasts (MEFs) with an hPSC medium and repeatedly freezing, thawing, and resuspending the culture until colonies appear. Colonies then need to be occasionally fragmented to encourage growth. With the iPSCs now prepared for differentiation, neuronal induction can occur once colonies are plated with a neural induction medium and then incubated for 8-12 days. After the incubation period, a neuroepithelial sheet will appear, which then needs to be fragmented by pipetting. The plated culture will then need to be put in a neural maintenance medium which will lead to the formation of neural rosettes. The neural rosettes will need to be centrifuged and then resuspended in medium frequently over 20 to 30 days for neurogenesis to occur. Differentiation into glutamatergic neurons will come with loss of expression of pluripotency genes and expression of transcription factors and proteins associated with excitatory glutamatergic neurons (vGlut1 and PSD-95). Shi et. al. explains that the efficiency of this method is dependent upon the pluripotent stem cells used in differentiation.
Methods to identify glutamatergic transformation: While the process detailed by Shi et. al. (2012) is certainly longer than that of Begum et. al. (2015), they both will lead to robust neuron populations differentiated into glutamatergic neurons. After differentiating the iPSCs, one can confirm the presence of glutamatergic neurons using immunofluorescence staining with the antibodies associated with this neuron type: vGlut1, PSD-95, and Homer1. If the cells viewed are tagged with the appropriate immunofluorescence stain, it can be confirmed that glutamatergic neurons were differentiated from the iPSC cultures.
2) GABAergic
a) Medium Spiny Neuron (MSN) Differentiation
The mechanism described by Begum et. al. (2015) above can also be applied to GABAergic neurons, with their appearance occurring a week after neuronal induction. Considering this method has already been described, the protocol will not be repeated in this section.
Grigor’eva’s method of differentiation: GABAergic differentiation can occur through various routes. Another possible method for GABAergic differentiation is using Grigor’eva et. al.’s (2020) protocol for a specific subtype of GABAergic neurons: medium spiny neurons (MSNs). Once iPSC confluency is at 70-80%, they should be plated in neuronal differentiation medium for 5 days. In the following 7 days, the neuronal differentiation medium should not include SB431542 inhibitor or dorsomorphin. At day 12, the neuroectodermal cells in the medium should be disaggregated and then replated with the same medium used previously but supplemented with ROCK inhibitor. Two days later, the medium should change again to a 1:1 ratio of the current medium:NeuroB (which includes B27 and recombinant human brain derived neurotrophic factor). The medium will transition fully to NeuroB on day 15 and cell culturing should form a monolayer that is maintained every 7-10 days. At this point, the precursor to MSNs should be fully formed, and in order to generate mature MSNs, the culture should be plated in a NeuroBC medium (similar to the prior medium, but now containing recombinant human CTNF) for 10-20 days, with the medium being refreshed every other day. At this point, MSNs should be fully generated and ready for functional use.
Methods to identify GABAergic MSN transformation: When testing to demonstrate the presence of GABAergic neurons, immunofluorescence staining followed by microscopy will be used to identify the neurons. Using GAD65, GAD67, anti-GAD6, and VGat antibody stains on the differentiated neurons, it should be relatively easy to identify the presence of GABAergic neurons (Begum et. al. 2015 & Zhang et. al. 2013). More specifically, to confirm the presence of MSNs, Grigor’eva et. al. (2020) tagged terminally differentiated MSNs with TUJ1, GABA, and NF200 antibodies. Upon visualization, these markers confirm the presence of MSNs differentiated from iPSCs. Neurons that exhibit immunofluorescence from the aforementioned antibodies can be confirmed as GABAergic neurons, and, in the case of Grigor’eva et. al. (2020), medium spiny neurons.
b) GABAergic Purkinje Differentiation
Begum’s method of differentiation and identification of transformation: Purkinje neurons, a subtype of inhibitory neurons located in the cerebellar cortex of vertebrates, can be differentiated using the same process by which glutamatergic and general GABAergic neurons can be differentiated, based on the findings of Begum et. al. (2015). Once the progenitors are cultured from the iPSCs and placed in neuronal maintenance medium (NMM), cerebellar Purkinje neurons form approximately 40-45 days later. Upon developing sufficient quantities of Purkinje neurons, immunofluorescence imaging can once again be used to confirm the presence of these neurons. Calbindin, the marker most tightly associated with Purkinje neurons, should be identified in the differentiated cultures during confocal microscopy.
c) GABAergic Parvalbumin-Positive Interneuron Differentiation
Maroof’s method of differentiation and identification of transformation: Maroof et al. (2013) demonstrated how parvalbumin-positive cortical interneurons can be differentiated through pluripotent stem cells using a mouse cortical coculture strategy. Human embryonic stem cells (hESCs) as well as iPSCs are placed in a differentiation medium consisting of knockout serum replacer, N2 medium for neural induction, and neurobasal medium +B27 and N2 supplements for neuronal differentiation. Following this, recombinant growth factors are introduced to promote proper differentiation. Using the coculture system allowed for rapid maturation of neurons and enabled the expression of markers associated with the cortical interneuron subtype. In order to confirm the presence of parvalbumin-positive interneurons, cells were colabeled with calbindin antibody, a calcium-binding protein co-expressed by parvalbumin-positive interneurons, and then analyzed with Operetta, an automated high content imaging system. This immunohistochemistry analysis confirms the presence of parvalbumin-positive interneurons because cells that indicate expression of calbindin indicates properly differentiated cells.
3) Dopaminergic
Xue’s method of differentiation: Xue et al. (2019) describes a method in which differentiation of midbrain dopaminergic neurons is accomplished in a five-day time period using iPSCs and mRNA coding for two proneural transcription factors, Atoh1 and Ngn2. The iPSCs are plated with growth-factor reduced Matrigel and then transfected with Atoh1 mRNA for 3 days and Ngn mRNA for 1 day. The culture medium consists of SHH, FGF8b, and DAPT. After the five-day period, the medium is changed to be a neuron medium consisting of neurobasal medium with B27 supplement, BDNF, GDNF, TGFbeta-3, cAMP, ascorbic acid, and DAPT. Regular changing of the medium is useful to remove unattached cells and promote maturation into midbrain dopaminergic neurons.
Methods to identify dopaminergic transformation: In order to confirm the presence of midbrain dopaminergic neurons, Xue et al. stained neurons with primary antibodies, LMX1A, DAT, TH, Nurr1, and GIRK2 and conducted immunofluorescence imaging to determine if the cultured cells expressed the proteins associated with dopaminergic neurons. Expression of these antibodies is indicative of a successful differentiation into midbrain dopaminergic neurons.
4) Serotonergic
Serotonergic neurons have been generated from iPSCs, utilizing a procedure that involves human fibroblasts. Xu et. al. (2016) and Vadodaria et. al. (2016) both successfully used this method to form functional serotonergic neurons that they explain are promising in the context of studying selective serotonin reuptake inhibitors (SSRIs) and serotonin-related mental disorders.
Vadodaria’s method of differentiation: Vadodaria et. al. (2016) demonstrated that human dermal fibroblasts can be used to generate iPSCs that can then be differentiated into fully functional serotonergic neurons. The fibroblasts undergo transduction with lentiviral particles for pLVXEtO,39 ASCL1/NGN2, and induced serotonergic neuron factors. Following an incubation of approximately 24 hours, the transgenic-induced neuron and induced serotonergic neuron-competent fibroblasts are transferred into a tetracycline-free FBS-containing medium. An ASCL1/NGN2-based protocol is used for direct neuronal transdifferentiation and then fibroblasts are plated with induced neuron conversion medium for three weeks. After this time period, the medium is changed to neural maturation medium to further mature the induced neurons. The entire process takes roughly 3-6 weeks, ending with cultures of serotonergic neurons.
Xu’s method of differentiation: In Xu et. al. (2016), serotonergic neurons are also induced from human fibroblasts, specifically MRC5, CCD-19Lu, and IMR90, that are maintained in DMEM. Cotransfecting 293FT cells with FUW-tetO-LoxP-cDN formed the lentivirus that would then be used to infect the fibroblasts to change their morphology to appear more like neurons. After 16 hours, the medium infected with lentivirus is replaced with new DMEM, followed by neural induction medium 24 hours later. Doxycycline is included in the medium for the first seven days and dorsomorphin and SB431542 are included on days 2 through 7. Until sufficient cultures are generated, the medium must be replenished every other day. The overall timeline, from plating to generation of cultures, is approximately 15 days.
Methods to identify serotonergic neuron transformation: Immunofluorescence imaging of the serotonergic cells created was used in both Xu et. al. (2016) and Vadodaria et. al. (2016) in order to confirm the identity of the cells cultured. The antibody, 5-HT, was used in both studies to stain the neurons and serve as an indicator of the presence of serotonergic neurons.
5) Cholinergic
Begum’s method of differentiation: Begum et. al.’s method of differentiation through neurospheres is also applicable to the generation of cholinergic neurons, although cholinergic neurons do have some key differences to their methods than other neuron types. Importantly, cholinergic neurons form in the NMM much faster than other neuron types; after approximately 19-27 days of being maintained in the medium cholinergic neurons appeared. This is notably faster than other subtypes of neurons, such as Purkinje neurons, which require 40-45 days in the NMM before generation. All steps leading up to the generation of cholinergic neurons based on the Begum et. al. (2015) protocol remain the same.
Takayama’s method of differentiation: Alternatively, Takayama et. al. (2020) also successfully generated cholinergic neurons from iPSCs using a stepwise chemical induction method. The iPSC lines must be maintained in a medium for up to three days to allow for the generation of embryonic bodies. These embryonic bodies are then cultured in knockout serum replacement for 13 days (with routine changing of the medium) before being plated in a new neuronal differentiation medium containing N2 medium, forskolin, ascorbic acid, recombinant human brain-derived neurotrophic factor, recombinant human glial-cell-derived neurotrophic factor, recombinant human nerve growth factor, and recombinant human neurotrophin 3. Recombinant human ciliary neurotrophic factor is also present in the medium and assists with the differentiation into parasympathetic neurons. The medium is changed every two weeks until cholinergic neuron colonies form. A key point to consider is the cholinergic neurons are derived from a specific neural crest; Takayama’s method utilizes a neural crest induction step followed by an autonomous specification step with restricted WNT signaling inhibition, sonic hedgehog signaling inhibition, and bone morphogenic protein signaling activation which helps guide the differentiation to specific lineages to ensure cholinergic neurons are derived.
Methods to identify cholinergic neuron transformation: Immunofluorescence imaging with antibody tagging is, once again, the most effective way to determine if the cultured cells were properly differentiated. Begum and Takayama explain that the two antibody tags that are most tightly linked to cholinergic expression are CHAT and nAchR. If the cells stained with these two antibodies exhibit immunofluorescence once imaged, it can be understood that they are cholinergic neurons that have successful expression. Confocal microscopy is often used to visualize the antibody stains (Begum et. al. 2015 and Takayama et. al. 2020).
6) Peripheral
a) Sensory Neurons
Chambers’ method of differentiation: The generation of sensory neurons from iPSCs has been successfully completed by Chambers et. al. (2012). Perhaps most exciting regarding Chambers’ method is that it is notably faster, yet still efficient in generating differentiated neurons. Using a combination of 5 small-molecule pathway inhibitors, Chambers’ was able to differentiate mature and effective neurons in approximately 10 days. The combination of SU5402, CHIR99021 and DAPT eliminates the expression of PAX6, which is the human neuroectoderm marker, and induces neuronal beta3-tubulin. Additionally, two other inhibitors, LDN-193189 and SB431542, replace Noggin and neutralize pluripotent stem cells. These 5 inhibitors, when used together (termed the LSB3i treatment), lead to efficient maturation of neurons generated from iPSCs using dual-SMAD inhibition. While all three major types of sensory neurons are generated (proprioceptors, mechanoreceptors, and nociceptors) more than half of those generated in Chambers’ study (approximately 60%) were nociceptors. Within 10 days, this process can be completed, and sensory neurons can be effectively harvested.
Methods to identify sensory neuron transformation: Sensory neuron generation can be confirmed through the use of immunofluorescence staining followed by microscopy. General sensory neurons can be identified by staining the cells with the following antibodies: BRN3A and ISL1 after day 11. Microscopy images showing stained cells expressing the antibody indicate that the cells generated are functioning sensory neurons. Since Chambers et. al. focused their studies specifically on nociceptor neurons, as they are the most plentiful form of sensory neurons derived from this method. Because of this, antibody stains specific to nociceptors can also be used to further confirm the subtype of sensory neuron derived. The following antibody tags can be used to identify nociceptor sensory neurons: RUNX1, RET, SCN10A, P2RX3, TPVR1. Positive expression of these antibodies in imaging indicates that the cells tagged are successfully differentiated nociceptor sensory neurons.
b) Autonomic Nervous System Neurons
Saito-Diaz’s method of differentiation: Postganglionic sympathetic neurons of the peripheral nervous system have been successfully differentiated from iPSCs in study of Saito-Diaz et. al. (2019). This protocol can be divided into key steps: developing neural crest cells, culturing as neural crest spheroids, and long-term differentiation. To form the neural crest cells, iPSCs are plated with a neural induction cocktail that activates and inhibits specific pathways necessary for neural crest cells. After approximately 11 days, the neural crest cells are isolated and fluorescence-activated cell sorting is used to purify them, keeping the SOX10/CD49D-positive cells to ultimately form neural crest spheroids. The spheroids should be suspended in neural crest spheroid medium and be prepared for differentiation by day 15 of the process. At this point, the spheroids are replated on poly-L-ornithine, laminin, and fibronectin (PO/LM/FN)-coated plates in autonomic neuron medium until at least day 19. After day 19, postganglionic neurons will be differentiated, however, it is still possible to replate the spheroids on PO/LM/FN-coated plates on day 20 with the autonomic neuron medium and further mature the neurons being generated.
Methods to identify autonomic nervous system postganglionic neuron transformation: Saito-Diaz references Zeltner et. al. (2016) when discussing the method for characterizing the differentiated neurons using their protocol. Zeltner et. al. (2016) clearly shows that immunostaining the cells and visualizing them with microscopy methods would allow for clear characterization of cells. Specifically, sympathetic autonomic neurons can be identified when marked with ASCL1, PHOX2A, SCG10 (STMN2), TH and DBH, all of which are antibody markers associated with autonomic neurons. Using this method, one can easily characterize the differentiated cells to ensure that they are sympathetic autonomic neurons.
IV. Conclusions
The differentiation protocols discussed above highlight the diversity of processes that can be utilized to achieve the neuron subtypes described. Importantly, the variety in methods suggests that different protocols may be better for different types of studies; a more time-sensitive study may, for example, benefit from shorter differentiation processes, such as described in Chambers et. al. (2012). Alternatively, methods that are more time-consuming, such as Begum et. al.’s (2015) 45-day process for differentiating Purkinje cells, should be assessed more analytically; is this lengthy process producing differentiated cells that are of significantly better quality than other faster methods described to justify the lengthy process? One of the major limitations in comparing the various differentiation protocols aforementioned is the fact that there is no standardized method to quantify the efficiency of these protocols in comparison to each other, as these papers are presenting their own differentiation methods. It is difficult to perform a cost-benefit analysis comparing these studies at this point due to the lack of uniformity in the data presented by each paper. Future studies hoping to finetune the efficiency and quality of differentiating iPSCs into neuronal subtypes could compare the various methods described here, focusing on assessing methods for differentiation into the same type of neuron subgroup as well as comparing across neuron subgroups in order to have a more holistic understanding of the merits of these various methods in comparison to one another. The use of immunofluorescence imaging was uniform across all methods to confirm the presence of the differentiated neurons, suggesting this is an accurate method to confidently confirm the identity of the cell that has been differentiated. By analyzing the commonalities across protocols, it is possible for researchers to determine which methods are most suitable to their specific goals.
There are no conflicts of interest associated with the production of this manuscript.
References
- Aoi T. 10th anniversary of iPS cells: the challenges that lie ahead. J Biochem. 2016;160(3):121- 129. doi:10.1093/jb/mvw044
- Begum AN, Guoynes C, Cho J, Hao J, Lutfy K, Hong Y. Rapid generation of sub-type, region- specific neurons and neural networks from human pluripotent stem cell-derived neurospheres. Stem Cell Res. 2015;15(3):731-741. doi:10.1016/j.scr.2015.10.014
- Chambers SM, Qi Y, Mica Y, et al. Combined small-molecule inhibition accelerates developmental timing and converts human pluripotent stem cells into nociceptors. Nat Biotechnol. 2012;30(7):715-720. Published 2012 Jul 1. doi:10.1038/nbt.2249
- Ferrari E, Cardinale A, Picconi B, Gardoni F. From cell lines to pluripotent stem cells for modelling Parkinson's Disease. J Neurosci Methods. 2020 Jul 1;340:108741. doi: 10.1016/j.jneumeth.2020.108741. Epub 2020 Apr 18. PMID: 32311374.
- Freel BA, Sheets JN, Francis KR. iPSC modeling of rare pediatric disorders. J Neurosci Methods. 2020 Feb 15;332:108533. doi: 10.1016/j.jneumeth.2019.108533. Epub 2019 Dec 4. Erratum in: J Neurosci Methods. 2020 Jun 1;339:108739. PMID:31811832; PMCID: PMC7310918.
- Grigor'eva EV, Malankhanova TB, Surumbayeva A, et al. Generation of GABAergic striatal neurons by a novel iPSC differentiation protocol enabling scalability and cryopreservation of progenitor cells. Cytotechnology. 2020;72(5):649-663. doi:10.1007/s10616-020-00406-7
- Liu G, David BT, Trawczynski M, Fessler RG. Advances in Pluripotent Stem Cells: History, Mechanisms, Technologies, and Applications. Stem Cell Rev Rep. 2020;16(1):3-32. doi:10.1007/s12015-019-09935-x
- Maroof AM, Keros S, Tyson JA, et al. Directed differentiation and functional maturation of cortical interneurons from human embryonic stem cells. Cell Stem Cell. 2013;12(5):559- 572. doi:10.1016/j.stem.2013.04.008
- Nakagawa M, Koyanagi M, Tanabe K, et al. Generation of induced pluripotent stem cells without Myc from mouse and human fibroblasts. Nat Biotechnol. 2008;26(1):101-106. doi:10.1038/nbt1374
- Nicholas CR, Chen J, Tang Y, et al. Functional maturation of hPSC-derived forebrain interneurons requires an extended timeline and mimics human neural development. Cell Stem Cell. 2013;12(5):573-586. doi:10.1016/j.stem.2013.04.005
- Saito-Diaz K, Wu HF, Zeltner N. Autonomic Neurons with Sympathetic Character Derived From Human Pluripotent Stem Cells. Curr Protoc Stem Cell Biol. 2019;49(1):e78. doi:10.1002/cpsc.78
- Shi Y, Inoue H, Wu JC, Yamanaka S. Induced pluripotent stem cell technology: a decade of progress. Nat Rev Drug Discov. 2017;16(2):115-130. doi:10.1038/nrd.2016.245
- Shi Y, Kirwan P, Livesey FJ. Directed differentiation of human pluripotent stem cells to cerebral cortex neurons and neural networks. Nat Protoc. 2012;7(10):1836-1846. doi:10.1038/nprot.2012.116
- Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126(4):663-676. doi:10.1016/j.cell.2006.07.024
- Takayama Y, Kushige H, Akagi Y, Suzuki Y, Kumagai Y, Kida YS. Selective Induction of Human Autonomic Neurons Enables Precise Control of Cardiomyocyte Beating. Sci Rep. 2020;10(1):9464. Published 2020 Jun 11. doi:10.1038/s41598-020-66303-3
- Vadodaria KC, Mertens J, Paquola A, et al. Generation of functional human serotonergic neurons from fibroblasts. Mol Psychiatry. 2016;21(1):49-61. doi:10.1038/mp.2015.161
- Xu Z, Jiang H, Zhong P, Yan Z, Chen S, Feng J. Direct conversion of human fibroblasts to induced serotonergic neurons. Mol Psychiatry. 2016;21(1):62-70. doi:10.1038/mp.2015.101
- Xue Y, Zhan X, Sun S, et al. Synthetic mRNAs Drive Highly Efficient iPS Cell Differentiation to Dopaminergic Neurons. Stem Cells Transl Med. 2019;8(2):112-123. doi:10.1002/sctm.18-0036
- Zhang Y, Pak C, Han Y, et al. Rapid single-step induction of functional neurons from human pluripotent stem cells. Neuron. 2013;78(5):785-798. doi:10.1016/j.neuron.2013.05.029
- Zeltner N, Fattahi F, Dubois NC, et al. Capturing the biology of disease severity in a PSC-based model of familial dysautonomia. Nat Med. 2016;22(12):1421-1427. doi:10.1038/nm.4220